
|
||


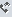



Mô hình trí tuệ nhân tạo mới giúp tìm ra các hoạt chất tiềm năng nhanh hơn hàng nghìn lần
Đăng lúc: Chủ nhật - 24/07/2022 17:39 - Người đăng bài viết: admin 
EquiBind có khả năng tìm ra các hoạt chất tiềm năng nhanh hơn 1,200 lần so với các mô hình trước đây. Ảnh: Hannes Stärk.
Vũ trụ của chúng ta chứa vô số phân tử, nhưng bao nhiêu trong số phân tử đó có tiềm năng để phát triển thành thuốc chữa bệnh? Hàng triệu phân tử? Hàng tỷ? Hàng nghìn tỷ? Câu trả lời là một con số khổng lồ: 10 mũ 60. Do vượt xa khả năng xử lý của các mô hình thiết kế thuốc đã có, quá trình phát triển thuốc cho các bệnh lây lan nhanh chóng như COVID-19 đòi hỏi phải có một khoảng thời gian nghiên cứu rất dài (để dễ hình dung, cần hiểu rằng Dải Ngân hà rộng lớn của chúng ta cũng mới chỉ có khoảng 100 tỷ ngôi sao mà thôi).
Trong nghiên cứu mới được trình bày tại Hội nghị Quốc tế về Học máy (ICML), các nhà nghiên cứu của Viện Công nghệ Massachusetts (MIT) đã phát triển một mô hình học sâu hình học có tên EquiBind, có khả năng xử lý nhanh hơn 1,200 lần so với QuickVina2-W – một trong những mô hình docking phân tử (kỹ thuật mô hình hóa giúp sàng lọc ảo các hợp chất trên máy tính) nhanh nhất hiện nay. EquiBind được phát triển dựa trên mô hình tiền thân có tên EquiDock – mô hình chuyên dùng để liên kết hai protein bằng kỹ thuật do Octavian-Eugen Ganea – đồng tác giả báo báo, nhà nghiên cứu tại Phòng thí nghiệm Khoa học Máy tính và Trí tuệ Nhân tạo và Jameel Clinic tại MIT – phát triển.
Trước khi bắt đầu phát triển thuốc, các nhà nghiên cứu phải chọn ra được những phân tử giống thuốc có tiềm năng liên kết (dock) chính xác với protein mục tiêu. Đây chính là quá trình tìm kiếm các hoạt chất tiềm năng (drug discovery). Sau khi liên kết thành công với protein, phối tử này sẽ có khả năng ngăn protein hoạt động. Nếu điều này xảy ra với một loại protein thiết yếu đối với vi khuẩn, phối tử ấy sẽ có thể tiêu diệt vi khuẩn và bảo vệ cơ thể con người.
Tuy nhiên, quá trình sàng lọc để tìm kiếm các hợp chất tiềm năng như vậy vô cùng tốn kém, có thể “ngốn” đến hàng tỷ đô-la và cần cả một thập kỷ để phát triển và thử nghiệm rồi mới có thể được cấp phép. Đó là chưa kể, 90% các loại thuốc trong nghiên cứu đều thất bại khi thử nghiệm trên cơ thể con người do không có tác dụng hoặc có quá nhiều tác dụng phụ. Cũng bởi vậy, những loại thuốc hiếm hoi thành công sẽ được các công ty thuốc nâng giá bán để bù đắp lại các chi phí nghiên cứu.
Hiện nay, hầu hết các mô hình tính toán hiện đại để tìm kiếm phân tử thuốc tiềm năng đều dựa nhiều vào việc lấy mẫu và các phương pháp như tính điểm, xếp hạng và tinh chỉnh để tìm ra được sự “phù hợp” nhất giữa phối tử và protein.
Tuy nhiên, Hannes Stärk (Khoa Kỹ thuật Điện và Khoa học Máy tính tại MIT) – tác giả chính của bài báo mới, cho rằng các phương pháp điển hình liên kết phối tử và protein như vậy giống như đang “cố gắng lắp một chiếc chìa khóa vào một ổ có nhiều lỗ khóa” – một cách tiếp cận tiêu tốn nhiều thời gian. Trong khi đó, mô hình EquiBind của nhóm nghiên cứu sẽ “liên kết mù” – dự đoán trực tiếp vị trí chính xác chỉ trong một bước mà không cần phải biết trước về protein đích.
Không giống như hầu hết các mô hình trước đây vốn đòi hỏi nhiều công sức để tìm ra vị trí thuận lợi cho phối tử trong protein, mô hình EquiBind đã tích hợp sẵn các suy luận hình học để có thể hiểu và khái quát hóa, đưa ra dự đoán chính xác hơn khi gặp dữ liệu mới hoặc chưa biết trước đó.
Kết quả nghiên cứu này đã nhanh chóng thu hút sự chú ý của các chuyên gia trong ngành, bao gồm cả Pat Walters – giám đốc dữ liệu của Relay Therapeutics. Walters đề nghị nhóm nghiên cứu thử mô hình của họ trên một loại thuốc và protein đã có và đang được sử dụng cho bệnh ung thư phổi, bệnh bạch cầu và các khối u đường tiêu hóa. Kết quả cho thấy, trong khi hầu hết các phương pháp docking truyền thống không thể liên kết thành công các phối tử với những protein ấy, thì EquiBind lại thành công.
“Với khả năng tận dụng thông tin từ hàng nghìn cấu trúc tinh thể khác nhau, phương pháp này sẽ có tiềm năng tác động đến lĩnh vực nghiên cứu thuốc theo nhiều cách mới”, Walters đánh giá.
Mỹ Hạnh dịch
Nguồn: https://techxplore.com/news/2022-07-artificial-intelligence-potential-drug-molecules.html
Nguồn tin: Tia Sáng
Những tin mới hơn
- AI sửa lỗi quy trình in 3D theo thời gian thực (23/08/2022)
- Lần đầu tiên bệnh di truyền về tim có thể được chữa khỏi (23/08/2022)
- Nghiên cứu sinh Nguyễn Thị Huệ đã bảo vệ thành công luận án tiến sĩ Vật Lý tại trường ĐH Tổng Hợp Warszawa, Ba Lan. (28/08/2022)
- Mặt tiền nhà nghiêng về phía trước với móc tời ở Amsterdam (02/09/2022)
- Biến thể BA.5 làm tăng tỷ lệ tái nhiễm (10/08/2022)
- Tế bào chuyển động trong niêm dịch nhanh hơn trong máu (06/08/2022)
- Đầu hồi đa dạng của những ngôi nhà ở Amsterdam (03/08/2022)
- Phát hiện giải pháp tạo siêu dẫn trong DNA có thể làm thay đổi công nghệ (06/08/2022)
- Toán học chứng tỏ có thể truyền thông lượng tử trong không gian liên sao (06/08/2022)
- Áp dụng các phương pháp xử lý dữ liệu xây dựng hệ tri thức chuyên gia Việt (28/07/2022)
Những tin cũ hơn
- Khủng hoảng đa dạng sinh học sẽ ảnh hưởng đến hàng tỉ người (24/07/2022)
- Giáo sư Ukraine và con đường đến 'giải Nobel Toán học 2022' (17/07/2022)
- BA.5 kháng vaccine cao gấp 4 lần biến chủng khác (17/07/2022)
- Thiết kế các bề mặt để sự sôi hiệu quả hơn (17/07/2022)
- Biến thể phụ BA.5 có gây bệnh nặng hơn các biến thể khác? (10/07/2022)
- Giải bình chọn sản phẩm Trí tuệ nhân tạo 2022 nhận hồ sơ (06/07/2022)
- Thạch Chính Lệ và nguồn gốc của Covid-19 (phần 4) (06/07/2022)
- Phát hiện gen giúp ngô thích nghi với môi trường cao nguyên (06/07/2022)
- Cỗ máy nano xâm nhập và tiêu diệt tế bào ung thư (20/06/2022)
- Phút chốc quên quên nhớ nhớ có thể là dấu hiệu của chứng sa sút trí tuệ (20/06/2022)






- Đang truy cập: 5
- Hôm nay: 1152
- Tháng hiện tại: 77929
- Tổng lượt truy cập: 25628511

 Đặt làm trang chủ
Đặt làm trang chủ  Liên hệ tòa soạn
Liên hệ tòa soạn Liên hệ quảng cáo
Liên hệ quảng cáo Đường dây nóng: (022 222 77 18)
Đường dây nóng: (022 222 77 18)
 Đăng ký thành viên CLB Lê Quý Đôn
Đăng ký thành viên CLB Lê Quý Đôn ,

Ý kiến bạn đọc